Odoo ERP for Medtech
In the highly regulated MedTech sector, achieving market maturity can be challenging due to stringent compliance requirements. Early investment in robust IT systems is crucial for ensuring compliance and facilitating growth. Traditional ERP solutions are often prohibitively expensive for emerging companies. Odoo Enterprise presents a cost-effective alternative, serving as a comprehensive, integrated part of a company's infrastructure. Here we will explore how Odoo Enterprise can help MedTech companies meet regulatory demands, adapt to market changes, and streamline operations.

Understanding MedTech
Here is a small crash course to get an overview over the basics!
What exactly is MedTech?
The MedTech sector encompasses everything to do with the development, manufacture, distribution and monitoring of medical device.
What is a medical device?
According to the ‘EU Medical Device Regulation (2017/745)’ (also known as MDR), a medical device is defined as an object of any kind, including software, primarily according to its area of application.
Examples of this are:
- Diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of diseases.
- Examination, replacement or modification of the anatomy or of a physiological or pathological process or condition and
- Obtaining information by the in-vitro examination of specimens derived from the human body, including organ, blood and tissue donations.
Medical devices must be intended for human use. Moreover, the mode of action is important, as medical devices act in or on the human body, but not pharmacologically, metabolically or immunologically.
However, there are some products for which this definition leads to difficulties in categorisation, such as wellness products, cosmetics or protective gear.
How are medical devices classified?
Depending on the risk associated with the use of the medical device, they are categorised into classes I - III, with I representing the lowest and III the highest risk. Depending on the class, softer or stricter regulations apply for quality or post-market surveillance.
- Class I medical devices do not harbour any methodological risks and are generally only used temporarily on patients. Typical examples include aids such as wheelchairs and walking aids.
- Class I r medical devices are reusable, the ‘r’ stands for ‘reusable’. This class includes reusable surgical instruments.
- Class II a medical devices involve a certain risk of use and may even be used in the body for a short period of time. Typical examples include contact lenses and hearing aids.
- Class II b medical devices have an increased methodological risk and are used over a long period of time. Class II b therefore includes ventilators, dialysis machines, dental implants and - surprisingly - condoms.
- Class III medical devices belong to the highest risk level. Their hazard potential is considerable, as they act directly on the heart or the circulatory system, for example. This becomes clear when you realise that we are talking about products such as pacemakers, stents or hip implants.
 Source: SPECTARIS - German Industry Association for Optics, Photonics, Analytical and Medical Technology
Source: SPECTARIS - German Industry Association for Optics, Photonics, Analytical and Medical Technology The MedTech sector
In terms of the global market share in the medical technology sector, 81% is accounted for by the 100 largest companies in the sector, with the largest 20 players accounting for 51% of the entire market (the next largest 80 companies therefore have a market share of 30% (data from 2019)).
Even if the market share of the remaining companies is very low, a large number of small and medium-sized companies are located in this sector in Germany. Specifically, around 95% of the 1,375 companies have fewer than 250 employees and almost 900 companies even have fewer than 50 employees. If micro (< 20 employees) and trading companies are included here, the number of companies rises to a total of 12,500. These small companies usually offer very specific products that are sold and used in a specialised niche.
The industry's export ratio is quite high at just under 66%. As a result, many companies are not only subject to local regulations (e.g. MDR), but also, for example, the strict North American regulations (e.g. FDA).
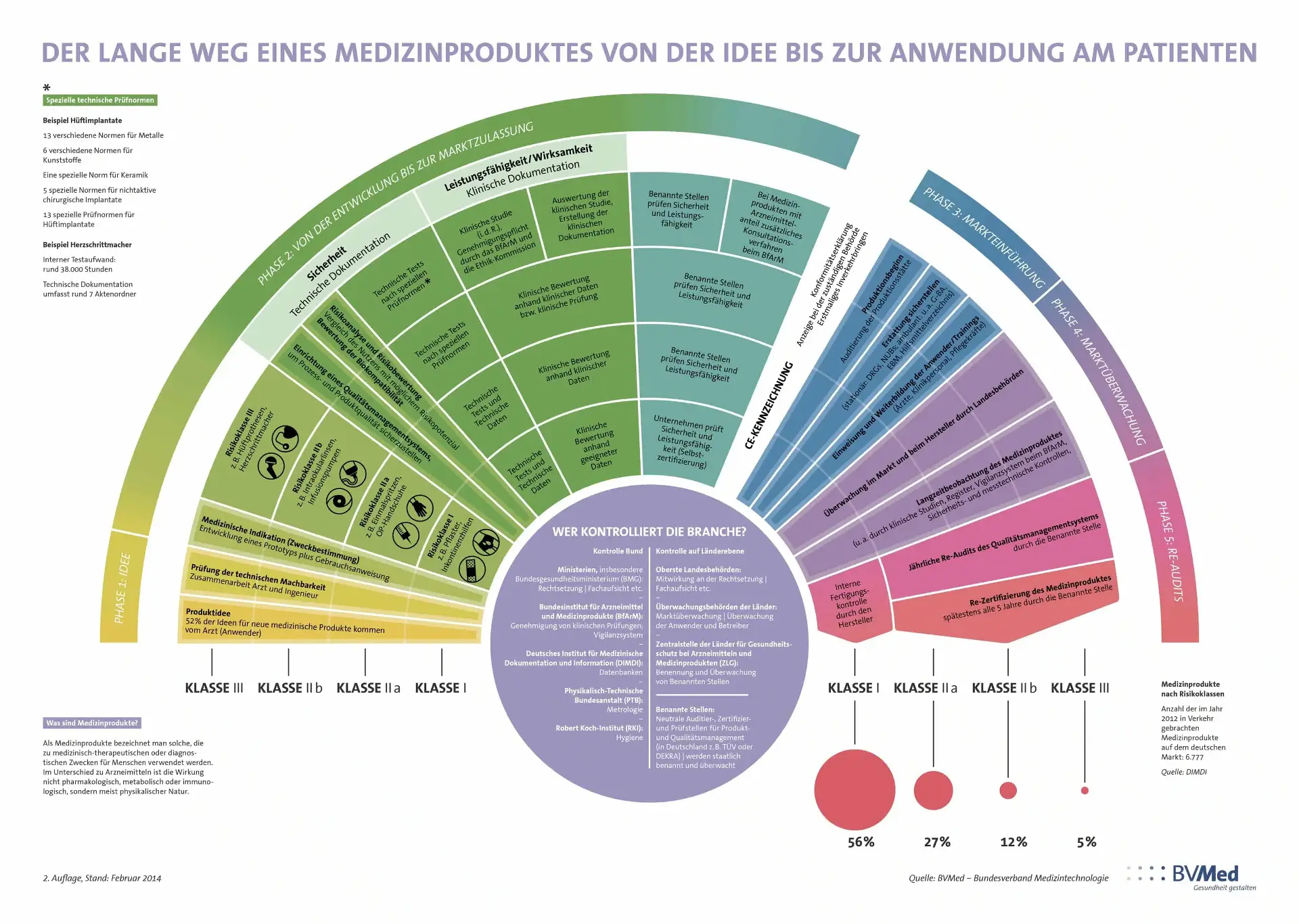
The biggest challenge in the MedTech sector are its strict regulations (MDR/FDA)
The biggest challenge is clearly the strict regulations - anyone who does not fulfil them is not allowed to sell the medical device! This is the most difficult step for small companies in particular. Time also plays a decisive role here, as these companies usually only generate stable income streams by selling their products. The following graphic provides an insight into the complexity of the regulations:
This shows that the idea for a new medical device is just the beginning. However, market authorisation is not the end of the story, as market surveillance and preparation for audits also require constant resources.
Regulations are set by the state and published in the form of (harmonised) standards or ordinances. Examples include EN ISO 13485 - which is an adapted version of the more general EN ISO 9001 for the MedTech sector - and the MDR. To ensure compliance, notified bodies such as TÜV-Süd are certified, which are then authorised by the state to carry out inspections. Such an inspection is called an ‘audit’ and can take place either announced or unannounced. If all rules have been complied with, the CE certificate is awarded and the product can be placed on the market or continue to be sold[4,5].
Even if the regulations on good manufacturing practice (GMP) strictly only apply to medicinal products, some companies decide to have their medical devices additionally certified for this purpose. This additional qualification is required by pharmaceutical manufacturers, among others, for the medical devices they use.

What does the MDR say?
The MDR describes various regulated areas that must be fulfilled, for example quality management (detailed in ISO 13485) or risk management.
Some of these points are discussed in detail here:
Risk management
According to the MDR, manufacturers are obliged to introduce an appropriate risk management system and apply it continuously.
Three aspects must be covered here:
- When processing complaints, for example, the type of complaint (case of damage, etc.) and whether it is a case of incorrect operation, etc. are queried. Appropriate workflows must be introduced here to ensure the necessary process reliability.
- The risk analysis and risk assessment is responsible for categorising the medical device into one of the above-mentioned classes. However, the process does not stop abruptly but continues after classification and market launch on the basis of customer data. This fulfils the requirements for post-market surveillance. The procedure must be carried out according to a specified method, for example the ‘Failure Mode and Effective Analysis’ (FMEA). Among other things, the following questions are asked here (EN ISO 14971):
- - How often does a particular failure occur in the first place?
- - How serious are the consequences?
- - What needs to be reported where and when?
- The so-called CAPA processes (Corrective And Preventive Action) are defined for the corrective and preventive measures. With the help of customer data, defects are recognised (at an early stage), documented and rectified accordingly. Corrective measures can range from revising the operating instructions to replacing defective parts at the customer's premises.
Traceability and UDIs
All medical devices must be traceable. However, it is not only the finished product that needs a unique serial or batch number, but also all the individual parts that are installed, which leads to increased requirements for manufacturers and suppliers.
In addition, according to the MDR, each finished medical device (UDI-DI) and each product-specific component (UDI-PI) must be labelled with a unique product identifier. These UDIs are approved before being placed on the market, applied to the product in a clearly visible and legible manner (e.g. by laser or etching) and then uploaded to an official database (GUDID/EUDAMED).
Quality management
EN ISO 13485 describes the precise specifications regarding the definition of and compliance with high quality standards and their documentation. In addition to detailed and complete documentation, particular emphasis is also placed on the management structure. For example, a responsible person is appointed who is responsible for compliance with the described quality and documentation. Examples from the area of quality are the special requirements for sterile medical devices or the specifications for the control of defective products.
Innovation
Once the medical device has been approved, it must also be kept up to date with the latest technology in order to regularly renew the CE certification. This is one of the reasons why most patents are registered in the MedTech sector every year - in figures, there were over 12,000 patents in 2016. This makes for a very fast-moving industry with relatively short product life cycles. Regular innovations are a prerequisite here, which is why MedTech companies generally invest around 9% of their resources in continuous research and development.

Discuss your MedTech ERP requirements personally with our experts.
Why does the MedTech sector need an ERP system?
For most small companies, the biggest hurdle is reaching market maturity, which also involves investment in IT infrastructure. However, costly solutions such as SAP are usually out of the question, as the steady stream of income is delayed. Manual documentation is time-consuming even for risk class I products and will no longer be practicable once the MDR comes into force in May 2021 at the latest.
In addition, unannounced audits can also be carried out by the notified bodies for some years now. With mainly manual data processing, weak points can quickly be found here, which in extreme cases can jeopardise the CE marking of the product. ERP systems ensure complete documentation that can be verified at any time, where nothing can be ‘forgotten’ or ‘left behind’.
In addition, the quality specifications must be adhered to, documented and traceable. ERP systems in which all data is linked together are used to prevent errors during any data transfers.
Automatic linking of customer feedback with the product and the customer also prevents errors in the necessary processing of complaints. By customising the ‘feedback classes’, the specified steps for reporting etc. can be triggered automatically.
In addition, there are companies that have to adapt their IT workflows due to the introduction of the MDR in order to continue to receive CE certification. Some processes were still possible manually or with less flexible ERP systems before this stricter regulation. According to a recent BVMed survey, 16% of medtech companies have not yet started preparing for the introduction of the MDR, so the demand for a fast IT solution for the increased standards will increase.
Odoo for MedTech by much. Consulting
MedTech as a user sector of ERP systems is a special case because it is subject to strict production requirements. These include the traceability of a product over the entire product life cycle including the audit trail, the dual control principle for some processes categorised as critical according to GxP or the time-critical delivery of goods that fall under the best-before date. In addition, the compliance guidelines of every medical technology company are constantly monitored.
An ERP system that is to prove compliance with the GAMP criteria during inspections must be certified in accordance with DIN EN ISO 13485.
For this purpose, much. Consulting has created a special Odoo Medtech solution that fulfils the prerequisites for implementing all of these requirements.
 Odoo for MedTech includes all the customisations needed to use Odoo optimally for medical technology as well.
Odoo for MedTech includes all the customisations needed to use Odoo optimally for medical technology as well.To optimise Odoo for the medtech sector, we have made various system adaptations:
Complaints
Complete complaints solution with various authorisations, automation and weekly status reports.
Product/devices
Each device/product is tracked by serial number:
- All serial numbers entered + repair-related changes
- Originally created parts list, production steps, version
- Service records
- Notes, tickets & complaints
- Warranty
Tracebility
Each incoming part is tracked by serial number and linked to the device, all quality checks are tracked.
Quality
Advanced quality checks and tracking for manufacturing
Maintenance
Maintenance tracking with questionnaire and changed parts linked to the device;
Software updates are also tracked with the affected machines
PLM
We track changes at ERP level (impact on BOM) that are created elsewhere. Manufacturing changes are approved in Odoo. An evaluation questionnaire is also attached to the change
Supplier
Supplier level & certifications are tracked
