Odoo ERP für Medizintechnik
Im stark regulierten Medizintechniksektor kann das Erreichen der Marktreife aufgrund der strengen Compliance-Anforderungen eine Herausforderung sein. Frühzeitige Investitionen in robuste IT-Systeme sind entscheidend, um Vorschriften einzuhalten und das Wachstum zu erleichtern. Traditionelle ERP-Lösungen sind für aufstrebende Unternehmen jedoch oft unerschwinglich. Odoo Enterprise ist eine kostengünstige Alternative, die als umfassender, integrierter Teil der Unternehmensinfrastruktur dient. Hier werden wir untersuchen, wie Odoo Enterprise Medizintechnikunternehmen dabei hilft, gesetzliche Anforderungen zu erfüllen, sich an Marktveränderungen anzupassen und Prozesse zu optimieren.

Medizintechnik
verstehen
Hier ein kleiner Crashkurs, um einen Überblick über die Grundlagen zu bekommen!
Was genau ist Medizintechnik?
Die Medizintechnik Branche beschreibt alles rund um die Entwicklung, Herstellung, den Vertrieb sowie die Überwachung von Medizinprodukten.
Was ist ein Medizinprodukt?
Hierbei hilft die Definition eines Medizinprodukts nach der ‘Verordnung über Medizinprodukte EU (2017/745)’ (auch MDR genannt), welche vereinfacht ausgedrückt Medizinprodukte als Gegenstände aller Art inklusive Software vor allem nach ihren Anwendungsgebieten einordnet.
Beispiele hierfür sind:
- Diagnose, Verhütung, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten
- Untersuchung, Ersatz oder Veränderung der Anatomie oder eines physiologischen oder pathologischen Vorgangs oder Zustands und
- Gewinnung von Informationen durch die In-vitro-Untersuchung von aus dem menschlichen Körper, auch aus Organ-, Blut- und Gewebespenden stammenden Proben.
Medizinprodukte müssen für den Menschen bestimmt sein. Außerdem ist das Wirkprinzip wichtig, denn Medizinprodukte wirken im oder am menschlichen Körper, jedoch nicht pharmakologisch, metabolisch oder immunologisch.
Hierbei gibt es jedoch einige Produkte bei denen diese Definition zu Schwierigkeiten bei der Einteilung führt wie beispielsweise Wellnessprodukte, Kosmetik oder persönliche Schutzausrüstung.
Wie werden Medizinprodukte klassifiziert?
Je nach dem welches Risiko mit dem Einsatz des Medizinprodukts einher geht werden diese in Klassen von I – III eingeteilt, wobei I das geringste und III das höchste Risiko darstellt. Je nach Klasse gelten weichere oder schärfere Regulationen für die Qualität oder die Überwachung nach dem Inverkehrbringen.
Aufgeschlüsselt gelten die folgenden Klassen:
- Medizinprodukte der Klasse I bergen keine methodischen Risiken und werden in der Regel nur vorübergehend am Patienten angewendet. Typische Beispiele sind unter anderem Hilfsmittel wie Rollstühle und Gehhilfen.
- Medizinprodukte der Klasse I r sind wiederverwendbar, das „r“ steht für „reusable“. Unter die Klasse fallen unter anderem wiederverwendbare chirurgische Instrumente.
- Medizinprodukte der Klasse II a beinhalten ein gewisses Anwendungsrisiko und werden gegebenenfalls sogar kurzzeitig im Körper angewendet. Typische Beispiele sind unter anderem Kontaktlinsen sowie Hörgeräte.
- Medizinprodukte der Klasse II b verfügen über ein erhöhtes methodisches Risiko und werden über einen langen Zeitraum angewendet. Zu Klasse II b gehören deshalb Beatmungsgeräte, Dialysegeräte, Dentalimplantate und – überraschenderweise auch Kondome. .
- Medizinprodukte der Klasse III gehören der höchsten Risikostufe an. Ihr Gefahrenpotenzial ist erheblich, da sie beispielsweise direkt am Herz oder dem Kreislaufsystem wirken. Deutlich wird dies, wenn man sich vor Augen führt, dass es sich um Produkte wie Herzschrittmacher, Stents oder Hüftimplantate handelt.
 Quelle: SPECTARIS – Deutscher Industrieverband für Optik, Photonik, Analysen- und Medizintechnik e.V.
Quelle: SPECTARIS – Deutscher Industrieverband für Optik, Photonik, Analysen- und Medizintechnik e.V.
Der Medizintechnik-Sektor
Bezogen auf den weltweiten Marktanteil im Medizintechnikbereich , fallen 81% auf die 100 größten Unternehmen der Sparte, wobei die größten 20 Player 51% des gesamten Marktes unter sich aufteilen (die nächstgrößten 80 Unternehmen haben also einen Marktanteil von 30% (Daten aus 2019)).
Auch wenn der Marktanteil der restlichen Unternehmen sehr gering ist, sind in Deutschland eine große Anzahl an kleinen und mittelständigen Unternehmen in diesem Bereich angesiedelt. Im Detail haben rund 95% der 1.375 Unternehmen weniger als 250 Mitarbeiter und knapp 900 Betriebe sogar weniger als 50 Mitarbeiter. Nimmt man hier noch Kleinst-(< 20 Beschäftigte) und Handelsbetriebe auf, steigt die Zahl der Unternehmen auf insgesamt 12.500. Diese kleinen Unternehmen bieten meist sehr spezifische Produkte an, welche in einer speziellen Nische verkauft und angewendet werden.
Die Exportquote der Branche ist mit knapp 66% recht hoch. Deshalb sind für viele Unternehmen nicht nur die lokalen Regularien (z.B. MDR), sondern auch beispielsweise die scharfen nordamerikanischen Vorschriften (z.B. der FDA) relevant..
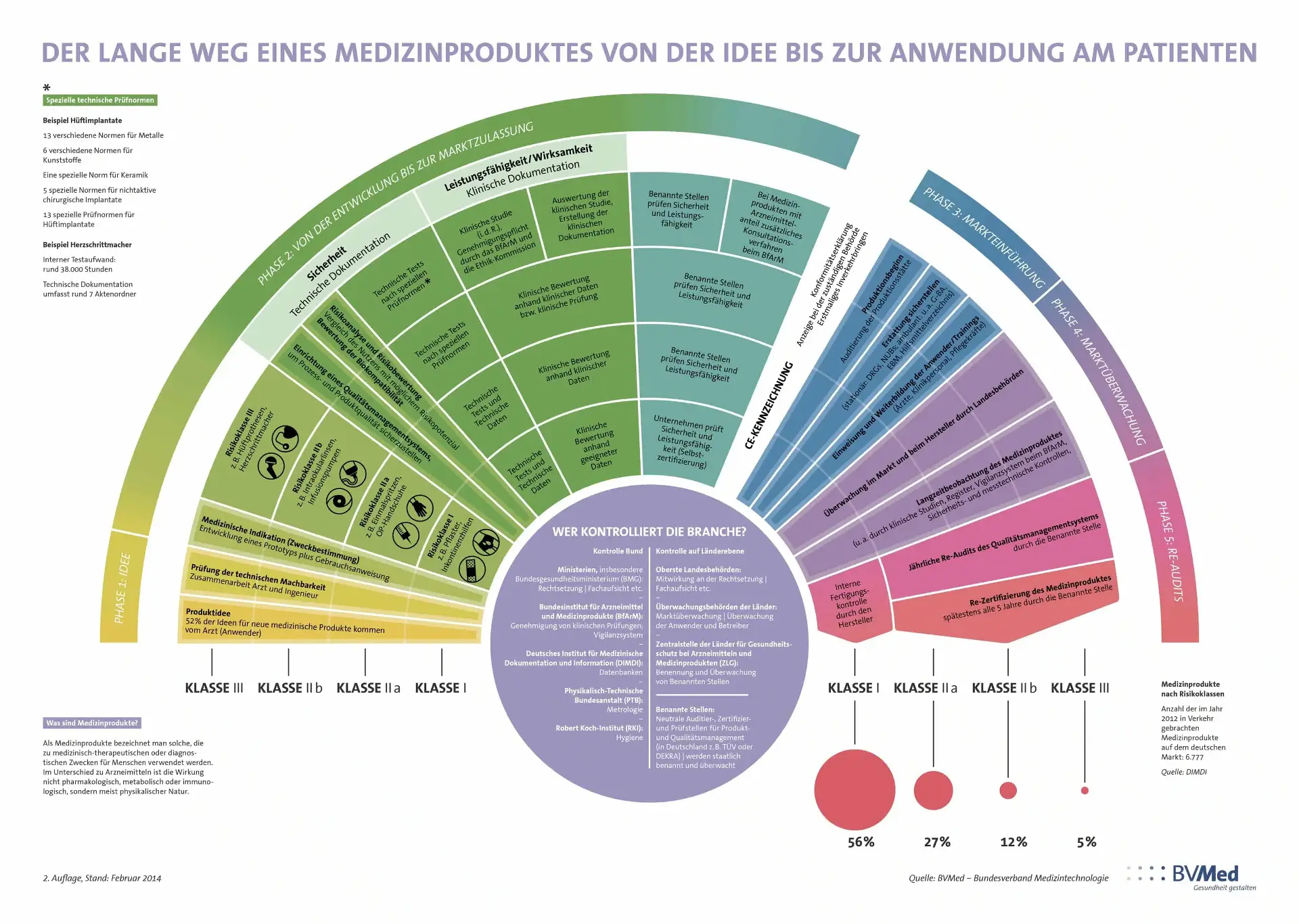
Die größte Herausforderung im medzintechnischen Bereich sind die strengen Vorschriften (MDR/FDA)
Als größte Herausforderung stechen eindeutig die strikten Regulierungen heraus – wer diese nicht erfüllt darf das Medizinprodukt nicht verkaufen! Besonders für kleine Unternehmen stellt dies den schwierigsten Schritt dar. Hier spielt auch die Zeit eine entscheidende Rolle, da diese Unternehmen meist erst durch den Verkauf ihrer Produkte stabile Einkommensströme generieren. Als Einblick in die Komplexität der Regulierungen soll die hiergezeigte Grafik dienen.
Hier wird gezeigt, dass die Idee für ein neues Medizinprodukt nur der Anfang ist. Jedoch ist auch mit der Marktzulassung nicht alles getan, denn auch die Marktüberwachung und die Vorbereitung auf Audits fordert konstant Ressourcen.
Regulierungen werden vom Staat vorgegeben und in Form von (harmonisierten) Normen oder Verordnungen publiziert. Beispiele hierfür sind die EN ISO 13485 – welche eine für den MedTech Sektor angepasste Version der allgemeineren EN ISO 9001 ist – und die MDR. Um die Einhaltung zu gewährleisten werden Benannte Stellen wie z.B. TÜV-Süd zertifiziert welche dann die staatliche Autorität zur Prüfung erhalten. Eine solche Prüfung wird „Audit“ genannt und kann entweder angekündigt oder unangekündigt stattfinden. Sind alle Regeln eingehalten worden wird das CE-Zertifikat vergeben und das Produkt kann auf den Markt gebracht bzw. weiterhin verkauft werden[4,5].

Was besagt die MDR?
Die MDR beschreibt verschiedene regulierte Bereiche welche erfüllt werden müssen, beispielsweise das Qualitätsmanagement (detailliert in ISO 13485) oder das Risikomanagement. Im folgenden wird auf einige dieser Punkte im Detail eingegangen..
Einige dieser Punkte werden hier im Detail erläutert:
Risikomanagement
Hersteller sind nach MDR verpflichtet ein entsprechendes Risikomanagementsystem einzuführen und dieses kontinuierlich anzuwenden.
Hierbei sind drei Aspekte abzudecken:
- Bei der Bearbeitung von Reklamationen, wird z.B. abgefragt um welche Art der Reklamation es sich handelt (Schadensfall etc.) und ob es sich um eine Fehlbedienung etc. handelt. Hier müssen entsprechende Arbeitsabläufe eingeführt werden um die nötige Prozesssicherheit zu gewährleisten.
- Die Risikoanalyse und Risikobewertung ist für die Einteilung des Medizinprodukts in eine der oben genannten Klassen verantwortlich. Der Prozess hört jedoch nicht abrupt auf sondern wird auch nach der Klassifizierung und der Markteinführung auf Basis der Kundendaten weitergeführt. Hierdurch werden die Anforderungen an die Marktüberwachung (Post Market Surveillance) erfüllt. Das Verfahren muss hier nach einer vorgegebenen Methode durchgeführt werden, beispielsweise der „Failure Mode and Effective Analysis“ (FMEA). Hier werden unter anderem die folgenden Fragen gestellt(EN ISO 14971):
- - Wie häufig tritt ein bestimmter Fehler überhaupt auf?
- - Wie schwerwiegend sind die Folgen?
- - Was muss wo und wann gemeldet werden?
- Die sogenannten CAPA-Prozesse (Corrective And Preventive Action) sind für die Korrektur- und Vorbeugemaßnahmen festgelegt. Hier werden mithilfe der Kundendaten Mängel (frühzeitig) erkannt, dokumentiert und entsprechend behoben. Korrekturmaßnahmen können hierbei weitläufig von einer Überarbeitung der Bedienungsanleitung bis zum Austausch von mangelhaften Teilen beim Kunden reichen.
Rückverfolgbarkeit und UDIs
Alle Medizinprodukte müssen rückverfolgbar sein. Jedoch braucht nicht nur das fertige Produkt eine eindeutige Serien- oder Chargennummer sondern auch alle eingebauten Einzelteile, was zu erhöhten Anforderungen für Hersteller und Zulieferer führt.
Zusätzlich muss nach MDR jedes fertige Medizinprodukt (UDI-DI) und jeder produktspezifische Bestandteil (UDI-PI) mit einer eindeutigen Produktkennung versehen werden. Diese UDIs werden vor dem Inverkehrbringen freigegeben, gut sichtbar und leserlich auf dem Produkt angebracht (z.B. über Laser oder Ätzen) und anschließend in eine behördliche Datenbank (GUDID/EUDAMED) hochgeladen.
Qualitätsmanagement
In der EN ISO 13485 sind die genauen Spezifikationen bezüglich der Festlegung und Einhaltung der hohen Qualitätsansprüche sowie deren Dokumentation beschrieben. Besonderer Fokus liegt hier neben der ausführlichen und lückenlosen Dokumentation auch auf der Managementstruktur. Beispielsweise wird eine verantwortliche Person benannt, welche für die Einhaltung der beschriebenen Qualität und Dokumentation verantwortlich ist. Beispiele aus dem Bereich der Qualität sind die besonderen Anforderungen an sterile Medizinprodukte oder die Vorgaben zur Lenkung von fehlerhaften Produkten.
Innovation
Ist das Medizinprodukt erstmal zugelassen, muss es auch auf dem Stand der Technik gehalten werden um die CE-Zertifizierung regelmäßig zu erneuern. Unter anderem deshalb werden im MedTech Bereich jährlich die meisten Patente angemeldet – in Zahlen waren es über 12.000 Patente im Jahr 2016. Dies sorgt für eine sehr schnelllebige Branche mit relativ kurzen Produktlebenszyklen. Hier regelmäßige Innovationen vorausgesetzt, weshalb MedTech Unternehmen in der Regel ca. 9% ihrer Ressourcen in die kontinuierliche Forschung und Entwicklung investieren.

Besprechen Sie Ihre Medtech ERP Anforderungen mit unseren Experten.
Wofür benötigt der MedTech Bereich ein ERP-System?
Für die meisten kleinen Unternehmen ist die größte Hürde das Erreichen der Marktreife, was auch mit Investitionen in die IT-Infrastruktur einher geht. Allerdings sind kostspielige Lösungen wie SAP hierbei meist außer Frage, da der stetige Einkommensstrom erst verspätet einsetzt. Eine händische Dokumentation ist selbst bei Produkten der Risikoklasse I aufwändig und spätestens mit dem Beginn der MDR im Mai 2021 nicht mehr praktikabel.
Zusätzlich können seit einigen Jahren auch unangekündigte Audits von den benannten Stellen durchgeführt werden. Bei einer hauptsächlich manuellen Datenverarbeitung können hier schnell Schwachstellen gefunden werden, welche im Extremfall die CE-Kennzeichnung des Produkts gefährden. ERP Systeme sorgen hier für eine jederzeit nachweisbare lückenlose Dokumentation wo nichts „vergessen“ oder „liegen geblieben“ sein kann.
Darüber hinaus müssen die Qualitätsspezifikationen eingehalten, dokumentiert und rückverfolgbar sein. Um hier Fehler bei eventuellen Datenübertragungen vorzubeugen, greifen ERP Systeme bei denen alle Daten miteinander verknüpft werden.
Auch eine automatische Verknüpfung einer Kundenrückmeldung mit dem Produkt sowie dem Kunden verhindert Fehler bei der erforderlichen Bearbeitung der Reklamationen. Durch angepasste Einteilung der „Rückmeldungsklassen“ können hier die vorgegebenen Schritte zur Meldung etc. automatisch angestoßen werden.
Darüberhinaus gibt es Unternehmen, welche aufgrund der Einführung der MDR ihre IT-Workflows anpassen müssen um weiterhin die CE-Zertifizierung zu erhalten. Manche Vorgänge waren vor dieser verschärften Vorschrift noch manuell oder mit weniger flexiblen ERP Systemen möglich. Da nach einer aktuellen Umfrage des BVMed 16% der MedTech Unternehmen noch nicht mit der Vorbereitung auf die MDR Einführung begonnen haben, wird die Nachfrage nach einer schnellen IT-Lösung für die erhöhten Standards steigen.
Odoo für MedTech von much. Consulting
Die Medizintechnik als Anwenderbranche von ERP-Systemen stellt insofern einen Sonderfall dar, als sie strengen Produktionsanforderungen unterliegt. Dazu gehören unter anderem die Rückverfolgbarkeit eines Produkts über den gesamten Produktlebenszyklus inklusive Audit Trail, das Vier-Augen-Prinzip für einige nach GxP als kritisch eingestufte Prozesse oder die zeitkritische Lieferung von Waren, die unter das MHD fallen. Darüber hinaus werden die Compliance-Richtlinien eines jeden Medizintechnikunternehmens ständig überwacht.
Ein ERP-System, das bei Inspektionen die Einhaltung der GAMP-Kriterien nachweisen soll, muss nach DIN EN ISO 13485 zertifiziert sein.
Hierfür hat much. Consulting eine spezielle Odoo Medtech-Lösung erstellt, die die Voraussetzungen zur Umsetzung all dieser Anforderungen erfüllt.
 Odoo Für MedTech beinhaltet alle Anpasssungen um Odoo auch in der Medizintechnik optimal verwenden zu können
Odoo Für MedTech beinhaltet alle Anpasssungen um Odoo auch in der Medizintechnik optimal verwenden zu können
So haben wir Odoo ERP für die Medizintechnikbranche optimiert:
Reklamationen
Vollständige Reklamationslösung mit verschiedenen Berechtigungen, Automatisierung und wöchentlichen Statusberichten.
Geräte/Produkte
Jedes Gerät/Produkt wird anhand der Seriennummer mitverfolgt:
- Alle eingegebenen Seriennummern + reparaturbedingte Änderungen
- Ursprünglich angelegte Stückliste, Fertigungsschritte, Version
- Serviceaufzeichnungen
- Notizen, Tickets & Reklamationen
- Garantie
Rückverfolgung
Jedes eingehende Teil wird anhand der Seriennummer nachverfolgt und mit dem Gerät verknüpft, alle Qualitätsprüfungen werden nachverfolgt.
Qualität
Fortgeschrittene Qualitätsprüfungen und -verfolgung für die Fertigung.
Wartung
Nachverfolgung der Wartung mit Fragebogen und geänderten Teilen, verbunden mit dem Gerät;
Software-Updates werden ebenfalls mit den betroffenen Maschinen nachverfolgt
PLM
Wir verfolgen Änderungen auf ERP-Ebene (Auswirkung auf Stückliste), die an anderer Stelle erstellt werden. Fertigungsänderungen werden in Odoo genehmigt. Der Änderung ist auch ein Bewertungsfragebogen beigefügt.
Lieferant
Lieferantenebene & Zertifizierungen werden verfolgt.
